|
|
|
|
|
Next: 9.3 The Hartree-Fock Approximation |
|
|
|
|
|
|
Next: 9.3 The Hartree-Fock Approximation |
|
Exact solutions in quantum mechanics are hard to come by. In almost all cases, approximation is needed. The Born-Oppenheimer approximation in particular is a key part of real-life quantum analysis of atoms and molecules and the like. The basic idea is that the uncertainty in the nuclear positions is too small to worry about when you are trying to find the wave function for the electrons. That was already assumed in the earlier approximate solutions for the hydrogen molecule and molecular ion. This section discusses the approximation, and how it can be used, in more depth.
The general problem to be discussed in this section is that of a number of electrons around a number of nuclei. You first need to know what is the true problem to be solved, and for that you need the Hamiltonian.
This discussion will be restricted to the strictly nonrelativistic
case. Corrections for relativistic effects on energy, including those
involving spin, can in principle be added later, though that is well
beyond the scope of this book. The physical problem to be addressed
is that there are a finite number ![]()
![]()
The electrons will be numbered using an index ![]() ,
,![]() .
.![]() ,
,![]()
![]() ,
,![]() ,
,![]() .
.![]()
![]()
Under the stated assumptions, the Hamiltonian of the system consists
of a number of contributions that will be looked at one by one. First
there is the kinetic energy of the electrons, the sum of the kinetic
energy operators of the individual electrons:
Similarly, there is the kinetic energy of the nuclei,
Next there is the potential energy due to the attraction of the ![]()
![]()
Next there is the potential energy due to the electron-electron
repulsions:
Finally, there is the potential energy due to the nucleus-nucleus
repulsions,
Solving the full quantum problem for this system of electrons and
nuclei exactly would involve finding the eigenfunctions ![]()
You might guess solving this problem is a tall order, and you would be perfectly right. It can only be done analytically for the very simplest case of one electron and one nucleus. That is the hydrogen atom solution, using an effective electron mass to include the nuclear motion. For any decent size system, an accurate numerical solution is a formidable task too.
The general idea of the Born-Oppenheimer approximation is simple. First note that the nuclei are thousands of times heavier than the electrons. A proton is almost two thousand times heavier than an electron, and that does not even count any neutrons in the nuclei.
So, if you take a look at the kinetic energy operators of the two,
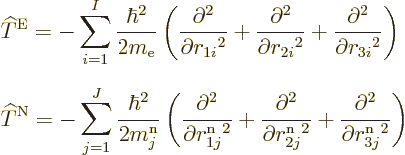
An alternative, and better, way of phrasing the assumption that
![]()
Assuming that the nuclei do not suffer from quantum uncertainty in
position is really equivalent to putting ![]()
![]()
![]()
![]()
![]()
That thought is the Born-Oppenheimer approximation in a nutshell.
Just do the electrons, assuming suitable positions for the nuclei a
priori. The solutions that you get doing so will be called ![]()
![]()
![]()
In application, it is usually most convenient to write the Hamiltonian
eigenvalue problem for the electron wave function as
Of course, after you compute your electron eigenfunctions, you want to
get something out of the results. Maybe you are looking for the
ground state of a molecule, like was done earlier for the hydrogen
molecule and molecular ion. In that case, the simplest approach is to
try out various nuclear positions and for each likely set of nuclear
positions compute the electronic ground state energy
![]() ,
,
For different assumed nuclear positions, you will get different values
for the electronic ground state energy, and the nuclear positions
corresponding to the actual ground state of the molecule will be the
ones for which the total energy is least:
This is what was used to solve the hydrogen molecule cases discussed
in earlier chapters; a computer program was written to print out the
energy ![]()
For more accurate work, the nominal ground state condition (9.13) above does have big limitations, so the next subsection discusses a more advanced approach.
Solving the wave function for electrons only, given positions of the nuclei is definitely a big simplification. But identifying the ground state as the position of the nuclei for which the electron energy plus nuclear repulsion energy is minimal is much less than ideal.
Such a procedure ignores the motion of the nuclei, so it is no use for figuring out any molecular dynamics beyond the ground state. And even for the ground state, it is really wrong to say that the nuclei are at the position of minimum energy, because the uncertainty principle does not allow precise positions for the nuclei.
Instead, the nuclei behave much like the particle in a harmonic oscillator. They are stuck in an electron blob that wants to push them to their nominal positions. But uncertainty does not allow that, and the wave function of the nuclei spreads out a bit around the nominal positions, adding both kinetic and potential energy to the molecule. One example effect of this “zero point energy” is to lower the required dissociation energy a bit from what you would expect otherwise.
It is not a big effect, maybe on the order of tenths of electron volts, compared to typical electron energies described in terms of multiple electron volts (and much more for the inner electrons in all but the lightest atoms.) But it is not as small as might be guessed based on the fact that the nuclei are at least thousands of times heavier than the electrons.
Moreover, though relatively small in energy, the motion of the nuclei
may actually be the one that is physically the important one. One
reason is that the electrons tend to get stuck in single energy
states. That may be because the differences between electron energy
levels tend to be so large compared to a typical unit ![]()
For example, the heat capacity of typical diatomic gases, like the hydrogen molecule or air under normal conditions, is not in any direct sense due to the electrons; it is kinetic energy of translation of the molecules plus a comparable energy due to angular momentum of the molecule; read, angular motion of the nuclei around their mutual center of gravity. The heat capacity of solids too is largely due to nuclear motion, as is the heat conduction of non metals.
For all those reasons, you would really, really, like to actually compute the motion of the nuclei, rather than just claim they are at fixed points. Does that mean that you need to go back and solve the combined wave function for the complete system of electrons plus nuclei anyway? Throw away the Born-Oppenheimer approximation results?
Fortunately, the answer is mostly no. It turns out that nature is
quite cooperative here, for a change. After you have done the
electronic structure computations for all relevant positions of the
nuclei, you can proceed with computing the motion of nuclei as a
separate problem. For example, if you are interested in the ground
state nuclear motion, it is governed by the Hamiltonian eigenvalue
problem
So, what is this ![]() ?
?![]()
![]()
![]()
![]() .
.![]()
And it does not necessarily have to be just to compute the ground state. You might want to study thermal motion or whatever. As long as the electrons are not kicked strongly enough to raise them to the next energy level, you can assume that they are in their ground state, even if the nuclei are not. The usual way to explain this is to say something like that the electrons “move so fast compared to the slow nuclei that they have all the time in the world to adjust themselves to whatever the electronic ground state is for the current nuclear positions.“
You might even decide to use classical molecular dynamics based on the
potential ![]()
So what if you are interested in what your molecule is doing when the
electrons are at an elevated energy level, instead of in their ground
state? Can you still do it? Sure. If the electrons are in an
elevated energy level ![]() ,
,![]() ,
,
Note that for a different value of ![]() ,
,![]()
![]() .
.adiabatic
normally
means processes sufficiently fast that heat transfer can be ignored.
So, some quantum physicists figured that it would be a good idea to
use the same term for quantum processes that are so slow that
quasi-equilibrium conditions persist throughout, and that have nothing
to do with heat transfer.
Of course, any approximation can fail. It is possible to get into
trouble solving your problem for the nuclei as explained above. The
difficulties arise if two electron energy levels, call them ![]()
![]() ,
,
You might now get alarmed, because for example the hydrogen molecular ion does have two different ground state solutions with the same energy. Its single electron can be in either the spin-up state or the spin down state, and it does not make any difference for the energy because the assumed Hamiltonian does not involve spin. In fact, all systems with an odd number of electrons will have a second solution with all spins reversed and the same energy {D.50}. There is no need to worry, though; these reversed-spin solutions go their own way and do not affect the validity of (9.14). It is spatial, rather than spin nonuniqueness that is a concern.
There is a derivation of the nuclear eigenvalue problem (9.14) in derivation {D.51}, showing what the ignored terms are and why they can usually be ignored.
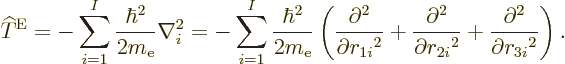
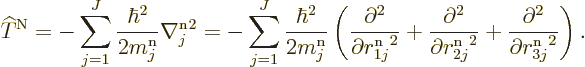
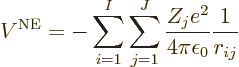
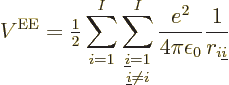
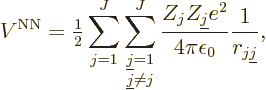
![\begin{displaymath}
\fbox{$\displaystyle
\left[{\widehat T}^{\rm E}+ V^{\rm NE}+ V^{\rm EE}\right] \psi^{\rm E}= E^{\rm E}\psi^{\rm E}
$} %
\end{displaymath}](img1987.gif)
![\begin{displaymath}
\fbox{$\displaystyle
\left[{\widehat T}^{\rm N}+ V^{\rm NN}+ E^{\rm E}_n\right] \psi^{\rm N}_n = E \psi^{\rm N}_n
$} %
\end{displaymath}](img1997.gif)