|
|
|
|
|
Next: 10.4 Covalent Materials |
|
Metals are unique in the sense that there is no true molecular equivalent to the way the atoms are bound together in metals. In a metal, the valence electrons are shared on crystal scales, rather than between pairs of atoms. This and subsequent sections will discuss what this really means in terms of quantum mechanics.
The simplest metal is lithium. Before examining solid lithium, first
consider once more the free lithium atom. Figure 10.4
gives a more realistic picture of the atom than the simplistic
analysis of chapter 5.9 did. The atom is really made up
of two tightly bound electrons in
states very close to the nucleus, plus a loosely bound third
![]()
valence
electron in an expansive
state. The core, consisting of the
nucleus and the two closely bound 1s electrons, resembles an helium
atom that has picked up an additional proton in its nucleus. It will
be referred to as the ![]()
atom core.
As far as the 2s
electron is concerned, this entire atom core is not that much
different from an hydrogen nucleus: it is compact and has a net charge
equivalent to one proton.
One obvious question is then why under normal circumstances lithium is a solid metal and hydrogen is a thin gas. The quantitative difference is that a single-charge core has a favorite distance at which it would like to hold its electron, the Bohr radius. In the hydrogen atom, the electron is about at the Bohr radius, and hydrogen holds onto it tightly. It is willing to share electrons with one other hydrogen atom, but after that, it is satisfied. It is not looking for any other hydrogen molecules to share electrons with; that would weaken the bond it already has. On the other hand, the 2s electron in the lithium atom is only loosely attached and readily given up or shared among multiple atoms.
Now consider solid lithium. A perfect lithium crystal would look as sketched in figure 10.5. The atom cores arrange themselves in a regular, repeating, pattern called the “ crystal structure.” As indicated in the figure by the thick red lines, you can think of the total crystal volume as consisting of many identical little cubes called “(unit) cells.”. There are atom cores at all eight corners of these cubes and there is an additional core in the center of the cubic cell. In solid mechanics, this arrangement of positions is referred to as the “body-centered cubic” (BCC) lattice. The crystal “basis” for lithium is a single lithium atom, (or atom core, really); if you put a single lithium atom at every point of the BCC lattice, you get the complete lithium crystal.
Around the atom cores, the 2s electrons form a fairly homogeneous
electron density distribution. In fact, the atom cores get close
enough together that a typical 2s electron is no closer to the atom
core to which it supposedly belongs
than to the
surrounding atom cores. Under such conditions, the model of the 2s
electrons being associated with any particular atom core is no
longer really meaningful. It is better to think of them as belonging
to the solid as a whole, moving freely through it like an electron
gas.
Under normal conditions, bulk lithium is “poly-crystalline,” meaning that it consists of many microscopically small crystals, or “grains,“ each with the above BCC structure. The “grain boundaries“ where different crystals meet are crucial to understand the mechanical properties of the material, but not so much to understand its electrical or heat properties, and their effects will be ignored. Only perfect crystals will be discussed.
Key Points
- Lithium can meaningfully be thought of as an atom core, with a net charge of one proton, and a 2s valence electron around it.
- In the solid, the cores arrange themselves into a
body-centered cubic(BCC) lattice.
- The 2s electrons form an
electron gasaround the cores.
- Normally the solid, like other solids, does not have the same crystal lattice throughout, but consists of microscopic grains, each crystalline, (i.e. with its lattice oriented its own way).
- The grain structure is critical for mechanical properties like strength and plasticity. But that is another book.
Even the quantum mechanics of a perfect crystal like the lithium one
described above is not very simple. So it is a good idea to start
with an even simpler crystal. The easiest example would be a
crystal
consisting of only two atoms, but two lithium
atoms do not make a lithium crystal, they make a lithium molecule.
Fortunately, there is a dirty trick to get a crystal
with only two atoms: assume that nature keeps repeating itself as
indicated in figure 10.6. Mathematically, this is called
using periodic boundary conditions.
It assumes that
after moving towards the left over a distance called the period, you
are back at the same point as you started, as if you are walking
around in a circle and the period is the circumference.
Of course, this is an outrageous assumption. If nature repeats itself at all, and that is doubtful at the time of this writing, it would be on a cosmological scale, not on the scale of two atoms. But the fact remains that if you make the assumption that nature repeats, the two-atom model gives a much better description of the mathematics of a true crystal than a two-atom molecule would. And if you add more and more atoms, the point where nature repeats itself moves further and further away from the typical atom, making it less and less of an issue for the local quantum mechanics.
Key Points
- Periodic boundary conditions are very artificial.
- Still, for crystal lattices, periodic boundary conditions often work very well.
- And nobody is going to put any real grain boundaries into any basic model of solids anyway.
To describe the energy eigenstates of the electrons in one-dimensional crystals in simple terms, a further assumption must be made: that the detailed interactions between the electrons can be ignored, except for the exclusion principle. Trying to correctly describe the complex interactions between the large numbers of electrons found in a macroscopic solid is simply impossible. And it is not really such a bad assumption as it may appear. In a metal, electron wave functions overlap greatly, and when they do, electrons see other electrons in all directions, and effects tend to cancel out. The equivalent in classical gravity is where you go down far below the surface of the earth. You would expect that gravity would become much more important now that you are surrounded by big amounts of mass at all sides. But they tend to cancel each other out, and gravity is actually reduced. Little gravity is left at the center of the earth. It is not recommended as a vacation spot anyway due to excessive pressure and temperature.
In any case, it will be assumed that for any single electron, the net
effect of the atom cores and smeared-out surrounding 2s electrons
produces a periodic potential that near every core resembles that of
an isolated core. In particular, if the atoms are spaced far apart,
the potential near each core is exactly the one of a free lithium atom
core. For an electron in this two atom crystal,
the
intuitive eigenfunctions would then be where it is around either the
first or the second core in the 2s state, (or rather, taking the
periodicity into account, around every first or every second core in
each period.) Alternatively, since these two states are equivalent,
quantum mechanics allows the electron to hedge its bets and to be
about each of the two cores at the same time with some probability.
But as soon as the atoms are close enough to start noticeably affecting each other, only two true energy eigenfunctions remain, and they are ones in which the electron is around both cores with equal probability. There is one eigenfunction that is exactly the same around both of the atom cores. This eigenfunction is sketched in figure 10.6; it is periodic from core to core, rather than merely from pair of cores to pair of cores. The second eigenfunction is the same from core to core except for a change of sign, call it a flip-flop eigenfunction. It is shown in figure 10.7. Since the grey-scale electron probability distribution only shows the magnitude of the wave function, it looks periodic from atom to atom, but the actual wave function is only the same after moving along two atoms.
To avoid the grey fading away, the shown wave functions have not been normalized; the darkness level is as if the 2s electrons of both the atoms are in that state.
As long as the atoms are far apart, the wave functions around each
atom closely resemble the isolated-atom ![]()
![]()
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,16...
...}}}
\put(136,105){\makebox(0,0)[t]{$\skew2\vec d$}}
\end{picture}
\end{figure}](img2155.gif) |
A one-dimensional crystal made up from four atoms is shown in figure
10.8. Now there are four energy eigenstates. The energy
eigenstate that is the same from atom to atom is still there, as is
the flip-flop one. But there is now also an energy eigenstate that
changes by a factor ![]()
![]()
![]() .
.
Key Points
- The electron energy eigenfunctions in a metal like lithium extend over the entire crystal.
- If the cores are relatively far apart, near each core the energy eigenfunction of an electron still resembles the 2s state of the free lithium atom.
- However, the magnitude near each core is of course much less, since the electron is spread out over the entire crystal.
- Also, from core to core, the wave function changes by a factor of magnitude one.
- The extreme cases are the fully periodic wave function that changes by a factor one (stays the same) from core to core, versus the flip-flop mode that changes sign completely from one core to the next.
- The other eigenfunctions change by an amount in between these two extremes from core to core.
There is a pattern to the wave functions of one-dimensional crystals
as discussed in the previous subsection. First of all, while the
spatial energy eigenfunctions of the crystal are different from those
of the individual atoms, their number is the same. Four free lithium
atoms would each have one ![]()
To be very precise, a similar thing is true of the inner 1s electrons.
But since the ![]()
The reason that the energy eigenfunctions take the form shown in
figure 10.8 is relatively simple. It follows from the
fact that the Hamiltonian commutes with the “translation
operator” that shifts the entire wave function over one atom
spacing ![]() .
.
Now commuting operators have a common set of eigenfunctions, so the
energy eigenfunctions can be taken to be also eigenfunctions of the
translation operator. The eigenvalue must have magnitude one, since
periodic wave functions cannot change in overall magnitude when
translated. So the eigenvalue describing the effect of an
atom-spacing translation on an energy eigenfunction can be written as
![]()
![]()
![]()
![]() .
.![]()
This can be verified for the example energy eigenfunctions shown in
figure 10.8. For the fully periodic eigenfunction ![]()
![]()
![]()
![]() :
:![]()
![]()
![]() ;
;![]()
![]()
![]() 1
1![]() .
.![]() .
.![]()
![]()
![]() ,
,![]()
![]()
![]()
![]() .
.
In general, for an ![]() -
-![]()
![]()
![]()
![]()
![]()
![]()
![]() .
.![]() :
:
Mathematically it is awkward to describe the energy eigenfunctions
piecewise, as figure 10.8 does. To arrive at a better
way, it is helpful first to replace the axial Cartesian coordinate ![]()
crystal coordinate
![]()
| (10.2) |
The advantage of the crystal coordinate ![]()
![]() -
-![]() .
.![]() .
.
This result is part of what is called “Floquet theory:”
If the Hamiltonian is periodic of periodIn physics, this result is known as “Bloch’s theorem,” and the Floquet-type wave function solutions are called “Bloch functions” orthe energy eigenfunctions are not in general periodic of period ,
but they do take the form of exponentials times functions that are periodic of period
Bloch waves,because Floquet was just a mathematician, and the physicists’ hero is Bloch, the physicist who succeeded in doing it too, half a century later. {N.20}.
The periodic part ![]()
![]()
![]()
![]()
![]()
![]() ,
,![]()
![]()
![]()
It is often more convenient to have the energy eigenfunctions in terms
of the Cartesian coordinate ![]()
![]() ,
,
As the previous subsection explained, the energy eigenfunctions in a
crystal take the form of a Floquet exponential times a periodic
function ![]() .
.
Writing the periodic function ![]()
![]() -
-![]()
![]()
![]()
![]()
![]() ,
,![]()
![]() ,
,
As the previous two subsections discussed, the energy eigenfunctions
in a one-dimensional crystal take the form of a Floquet exponential
![]()
![]() .
.![]() .
.![]() -
-
The Fourier ![]()
![]()
![]()
![]()
![]()
![]()
reciprocal lattice.
The spacing of the reciprocal
lattice, ![]()
![]()
![]()
![]()
![]() ,
,![]()
![]()
The Floquet ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() ,
,![]()
![]()
To be precise, the Floquet ![]()
![]()
![]() ,
,![]()
![]() .
.![]()
![]()
![]()
![]()
The first Brillouin zone are the points that are closest to the origin
on the ![]() -
-![]()
![]()
![]()
![]() ,
,![]()
Valence band. Conduction band. Band gap. Crystal. Lattice. Basis. Unit cell. Primitive vector. Bloch wave. Fourier analysis. Reciprocal lattice. Brillouin zones. These are the jargon of solid mechanics; now they have all been defined. (Though certainly not fully discussed.) But jargon is not physics. The physically interesting question is what are the energy levels of the energy eigenfunctions.
For the two-atom crystal of figures 10.6 and
10.7, the answer is much like that for the hydrogen
molecular ion of chapter 4.6 and hydrogen molecule of
chapter 5.2. In particular, when the atom cores are far
apart, the ![]()
![]() .
.![]()
When the distance ![]()
Next consider the case of a four-atom crystal, as shown in the second graph of figure 10.10. The fully periodic and flip flop states are unchanged, and so are their energies. But there are now two additional states. Unlike the fully periodic state, these new states vary from atom, but less rapidly than the flip flop mode. As you would then guess, their energy is somewhere in between that of the fully periodic and flip-flop states. Since the two new states have equal energy, it is shown as a double line in 10.10. The third graph in that figure shows the energy levels of an 8 atom crystal, and the final graph that of a 24 atom crystal. When the number of atoms increases, the energy levels become denser and denser. By the time you reach a one hundredth of an inch, one-million atom one-dimensional crystal, you can safely assume that the energy levels within the band have a continuous, rather than discrete distribution.
Now recall that the Pauli exclusion principle allows up to two electrons in a single spatial energy state. Since there are an equal number of spatial states and electrons, that means that the electrons can pair up in the lowest half of the states. The upper states will then be unoccupied. Further, the actual separation distance between the atoms will be the one for which the total energy of the crystal is smallest. The energy spectrum at this actual separation distance is found inside the vanishingly narrow vertical frame in the rightmost graph of figure 10.10. It shows that lithium forms a metal with a partially-filled band.
The partially filled band means that lithium conducts electricity well. As was already discussed earlier in chapter 6.20, an applied voltage does not affect the band structure at a given location. For an applied voltage to do that, it would have to drop an amount comparable to volts per atom. The current that would flow in a metal under such a voltage would vaporize the metal instantly. Current occurs because electrons get excited to states of slightly higher energy that produce motion in a preferential direction.
The explanation of electrical conduction in metals given in the previous subsection is incomplete. It incorrectly seems to show that beryllium, (and similarly other metals of valence two,) is an insulator. Two valence electrons per atom will completely fill up all 2s states. With all states filled, there would be no possibility to excite electrons to states of slightly higher energy with a preferential direction of motion. There would be no such states. All states would be red in figure 10.10, so nothing could change.
What is missing is consideration of the 2p atom states. When the atoms are far enough apart not to affect each other, the 2p energy levels are a bit higher than the 2s ones and not involved. However, as figure 10.11 shows, when the atom spacing decreases to the actual one in a crystal, the widening bands merge together. With this influx of 300% more states, valence-two metals have plenty of free states to excite electrons to. Beryllium is actually a better conductor than lithium.
Hydrogen is a more complicated story. Solid hydrogen consists of
molecules and the attractions between different molecules are weak.
The proper model of hydrogen is not a series of equally spaced atoms,
but a series of pairs of atoms joined into molecules, and with wide
gaps between the molecules. When the two atoms in a single molecule
are brought together, the energy varies with distance between the
atoms much like the left graph in figure 10.10. The wave
function that is the same for the two atoms in the current simple
model corresponds to the normal covalent bond in which the electrons
are symmetrically shared; the flip-flop function that changes sign
describes the anti-bonding
state in which the two
electrons are antisymmetrically shared. In the ground state, both
electrons go into the state corresponding to the covalent bond, and
the anti-bonding state stays empty. For multiple molecules, each of
the two states turns into a band, but since the interactions between
the molecules are weak, these two bands do not fan out much. So the
energy spectrum of solid hydrogen remains much like the left graph in
figure 10.10, with the bottom curve becoming a filled band
and the top curve an empty one. An equivalent way to think of this is
that the 1s energy level of hydrogen does not fan out into a single
band like the 2s level of lithium, but into two half bands, since
there are two spacings involved; the spacing between the atoms in a
molecule and the spacing between molecules. In any case, because of
the band gap energy required to reach the empty upper half 1s band,
hydrogen is an insulator.
The ideas of the previous subsections generalize towards three-dimensional crystals in a relatively straightforward way.
As the lithium crystal of figure 10.12 illustrates, in a
three-dimensional crystal there are three “primitive translation vectors.” The three-dimensional Cartesian
position ![]()
crystal coordinates
Note that the vectors ![]()
![]()
cubic unit cell
defined earlier in figure
10.5. However, ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Despite this requirement, there are still many ways of choosing the primitive translation vectors other than the one shown in figure 10.12. The usual way is to choose all three to extend towards adjacent cube centers. However, then it gets more difficult to see that no lattice point is missed when stepping around with them.
The parallelepiped shown in figure 10.12, with sides given by the primitive translation vectors, is called the “primitive cell.” It is the smallest building block that can be stacked together to form the total crystal. The cubic unit cell from figure 10.5 is not a primitive cell since it has twice the volume. The cubic unit cell is instead called the “conventional cell.”
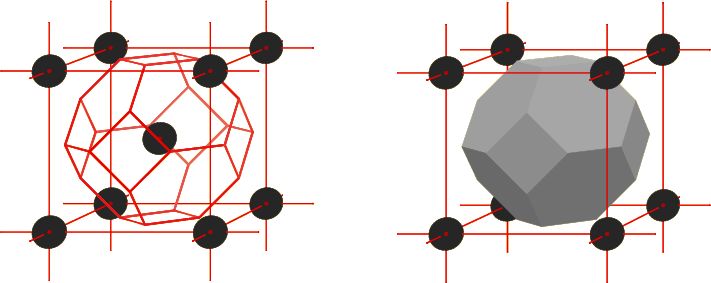
Since the primitive vectors are not unique, the primitive cell they define is not either. These primitive cells are purely mathematical quantities; an arbitrary choice for the smallest single volume element from which the total crystal volume can be build up. The question suggests itself whether it would not be possible to define a primitive cell that has some physical meaning; whose definition is unique, rather than arbitrary. The answer is yes, and the unambiguously defined primitive cell is called the “Wigner-Seitz cell.” The Wigner-Seitz cell around a lattice point is the vicinity of locations that are closer to that lattice point than to any other lattice point.
Figure 10.13 shows the Wigner-Seitz cell of the BCC lattice. To the left, it is shown as a wire frame, and to the right as an opaque volume element. To put it within context, the atom around which this Wigner-Seitz cell is centered was also put in the center of a conventional cubic unit cell. Note how the Wigner-Seitz primitive cell is much more spherical than the parallelepiped-shaped primitive cell shown in figure 10.12. The outside surface of the Wigner-Seitz cell consists of hexagonal planes on which the points are just on the verge of getting closer to a corner atom of the conventional unit cell than to the center atom, and of squares on which the points are just on the verge of getting closer to the center atom of an adjacent conventional unit cell. The squares are located within the faces of the conventional unit cell.
The reason that the entire crystal volume can be build up from Wigner-Seitz cells is simple: every point must be closest to some lattice point, so it must be in some Wigner-Seitz cell. When a point is equally close to two nearest lattice points, it is on the boundary where adjacent Wigner-Seitz cells meet.
Turning to the energy eigenfunctions, they can now be taken to be
eigenfunctions of three translation operators; they will change by
some factor ![]()
![]() ,
,![]()
![]() ,
,![]()
![]() .
.
It is again often convenient to write the Floquet exponential in terms
of normal Cartesian coordinates. To do so, note that the relation
giving the physical position ![]()
![]() ,
,![]() ,
,![]() ,
,
If you do not know linear algebra, it can be done geometrically: if
you dot the expression for ![]()
![]()
![]()
![]() ,
,![]() ;
;
If the expressions for the crystal coordinates are substituted into
the exponential part of the Bloch functions, the result is
Just like for the one-dimensional case, the periodic function
![]()
And now note the beautiful symmetry in the relations
(10.8) between the primitive vectors ![]() ,
,![]() ,
,![]()
![]() ,
,![]() ,
,![]()
![]() ,
,![]() ,
,![]()
![]() ,
,![]() ,
,![]() ,
,![]() ,
,![]() ,
,![]()
![]() ,
,![]() ,
,![]() .
.reciprocal
in reciprocal lattice comes from.
Lithium and NaCl borrow each other’s lattice to serve as their
lattice of wave number vectors.
Finally, how about the definition of the “Brillouin zones” in three dimensions? In particular, how about
the first Brillouin zone to which you often prefer to move the Floquet
wave number vector ![]() ?
?![]() ,
,![]() ,
,![]()
![]()
![]()
Solid state physicists may tell you that the other Brillouin zones are also reciprocal lattice Wigner-Seitz cells, [29, p. 38], but if you look closer at what they are actually doing, the higher zones consist of fragments of reciprocal lattice Wigner-Seitz cells that can be assembled together to produce a Wigner-Seitz cell shape. Like for the one-dimensional crystal, the second zone are again the points that are second closest to the origin, etcetera.
The boundaries of the Brillouin zone fragments are now planes called “Bragg planes.” Each is a perpendicular bisector of a lattice point and the origin. That is so because the locations where points stop being first/, second/, third/, ...closest to the origin and become first/, second/, third/, ...closest to some other reciprocal lattice point must be on the bisector between that lattice point and the origin. Sections 10.5.1 and 10.6 will give Bragg planes and Brillouin zones for a simple cubic lattice.
The qualitative story for the valence electron energy levels is the same in three dimensions as in one. Sections 10.5 and 10.6 will look a bit closer at them quantitatively.

![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(400,27...
... \put(0,0){\makebox(0,0)[b]{\epsffile{lithium.eps}}}
\end{picture}
\end{figure}](img2152.gif)
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,12...
...ebox(0,0)[bl]{${\left\vert 2s\right\rangle}^{(1)}$}}
\end{picture}
\end{figure}](img2153.gif)
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,12...
...box(0,0)[bl]{$-{\left\vert 2s\right\rangle}^{(2)}$}}
\end{picture}
\end{figure}](img2154.gif)
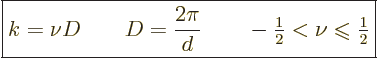
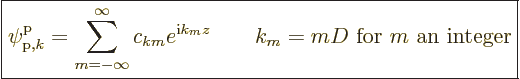
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(404,60...
...akebox(0,0)[b]{3}}
\put(76,20){\makebox(0,0)[b]{3}}
\end{picture}
\end{figure}](img2180.gif)
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,95...
...}}
\put(-183,31){\makebox(0,0)[bl]{fully periodic}}
\end{picture}
\end{figure}](img2185.gif)
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,15...
...x(0,0)[l]{2s}}
\put(76,117.5){\makebox(0,0)[l]{2p}}
\end{picture}
\end{figure}](img2186.gif)
![\begin{figure}\centering
\setlength{\unitlength}{1pt}
\begin{picture}(405,14...
...(0,1){4}}
\put(69,51){\makebox(0,0)[b]{$\vec d_3$}}
\end{picture}
\end{figure}](img2187.gif)
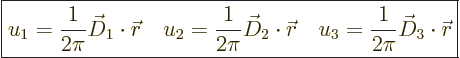
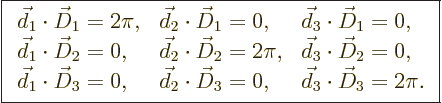
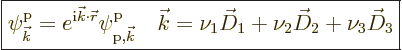
![\begin{displaymath}
\fbox{$\displaystyle
\begin{array}[b]{c}
\displaystyle
\...
..., $m_2$, and $m_3$\ integers}\strut^{\strut}
\end{array} $} %
\end{displaymath}](img2207.gif)